
一、常见的染色体非整倍体疾病
染色体非整倍体疾病是染色体疾病中发病率最高的一类。最常见的染色体非整倍体疾病有唐氏综合征(T21)、爱德华氏综合征(T18)和帕陶氏综合征(T13)。染色体疾病的发病原因至今不明,每一位准妈妈都有可能生育患有染色体疾病的宝宝。准妈妈孕龄越大,生育患病宝宝的可能性越大。
染色体疾病目前没有预防的办法,只能通过孕期检查来阻止患病宝宝的出生,所以在孕期对宝宝进行染色体疾病的检查对于每位准妈妈来说都非常重要。
| 疾病种类 | 异常区域 | 发病率 | 临床特征 |
|---|---|---|---|
| T21(唐氏综合征) | 21号染色体 | 1/750 | 特殊面容、多发畸形、生长发育迟缓、智力障碍,寿命减少 |
| T18(爱德华氏综合征) | 18号染色体 | 1/3500—1/8000 | 先天性心脏病,外表及多器官严重畸形,发育迟缓,40%存活至1个月,5%存活至1岁,1%存活至10岁 |
| T13(帕陶氏综合征) | 13号染色体 | 1/25000 | 95%以上会死于子宫内。半数以上的患者会有全前脑畸形,常合并先天性心脏病,活产寿命4-6个月 |
| 特纳综合征(45,X) | 性染色体 | 1/2500 女性新生儿 | 身材矮小,生殖器与第二性征发育不全,智力发育程度不一 |
| 克氏综合征(47,XXY) | 性染色体 | 1/500-1/1000 男性新生儿 | 体型较高,双侧睾丸较小,两侧乳房肥大,不育或性功能低下,智力发育正常或略低 |
| 超雄综合征(47,XYY) | 性染色体 | 1/1000 男性新生儿 | 通常躯体外形特征异常不显著,大多数性发育正常,可孕育下一代。可能存在学习障碍、语言发育迟缓,以及延迟的运动能力、肌张力低下、手震或其他不自主运动等 |
| 超雌综合征(47,XXX) | 性染色体 | 1/1000 女性新生儿 | 一般外表正常,躯体外形特征不显著。大多数性发育正常,可孕育下一代。可能存在学习障碍、语言发育迟缓,以及延迟的运动能力、肌张力低下等(47,XXY) |
折翼的天使——唐氏综合征患儿
有这样一群孩子,他们可爱,他们天真,总是用微笑去面对他人或善意或冷漠的眼关。相似的国际脸却泄露了他们的与众不同,他们就是唐氏综合征患者,一群降临人间的“折翼天使”。唐氏综合征,又称21三体综合征、先天愚型或Down综合征,是由于21号染色体三体而引起的先天畸形,发病率1/800-1/1000,是临床中最为常见的一种染色体疾病。
唐氏综合征患儿主要临床表现有:◆ 特殊面容,如眼距宽、鼻根低平,眼裂小,眼外侧上斜等
◆ 身材矮小、头围小于正常、骨骼发育严重滞后
◆ 智力低下、动作发育和性发育延迟
◆ 常伴有先天性心脏病、免疫功能低下等其他异常
唐氏综合征患儿的生育风险与孕妇年龄呈显著的相关性,孕妇年龄越大,生育唐氏综合征患儿的风险就越高。据统计,20-24岁孕妇生育唐氏综合征患儿的风险为1/1562,35-39岁的生育风险为1/214,而45岁以上则为1/19。不过,约80%的唐氏患儿由35岁以下的妇女生育,这主要是由于年轻妇女的生育率较高所导致。
目前唐氏综合征尚无有效的治疗手段,因而重视唐氏综合征的产前筛查与诊断便显得尤为重要。
二、不罕见的罕见病——染色体微缺失微重复综合征
染色体微缺失微重复是除染色体非整倍体疾病之外的另一大类新生儿出生缺陷,发病率1/4,000 -1/200,000不等,缺失和重复片段较为微小,通常被产前诊断漏检。有数据显示,大多数染色体微缺失微重复疾病为新发突变,并且发病风险与年龄无显著相关性。致病或可能致病的染色体微缺失微重复的发生率为1.7%,且再发风险很高。
染色体微缺失微重复患儿多数能正常存活,且出生后表现为不同程度的躯体或发育异常。以Digeorge综合征为例,其发病原因是由于22q11区段缺失2.5Mb区段导致。患儿多表现为先天性心脏病、上颚、胃肠、泌尿生殖系统发育异常、免疫缺陷、发育迟滞、认知障碍等临床表型。虽然小部分Digeorge综合征可以通过超声发现,但绝大多数仍会被漏掉。一项临床研究表明,每68个先心病患儿中就有一个患有Digeorge综合征。该综合征在新生儿中的发病率为1/1000-1/4000,比18三体综合征1/3500-1/7000的发病率还高。
| 22q11.2 deletion 综合征(含Digeorge综合征) | 1/4000 | 心脏病,血小板异常,面部特征异常,低血钙,进食困难,肾功能异常,发育语言迟缓,学习能力低下,免疫系统障碍,注意力缺陷多动障碍 |
| 1p36 deletion 综合征 | 1/5000-1/10000 | 特殊面容、视听障碍、心脏结构异常、癫痫、肌张力低下、多发畸形。生长迟缓、严重语言发展迟缓、情绪不易控制,寿命多数正常 |
| 2q33.1 deletion | 罕见 | 综合征特殊面容、癫痫、关节韧带松弛等症状。生长迟缓、严重语言发展迟缓、喂食困难、行为过动、躁动等症状 |
| Cri-Du-Chat 综合征 | 1/20000-1/50000 | 婴幼儿时期的哭声似小猫叫、特殊面容、肌张力低。生长迟缓、行为过度活跃、侵略、暴怒、重复动作等症,少数可活至成年 |
| Langer-Giedion 综合征 | 罕见 | 毛发稀疏、皮肤松弛、多发性骨疣、小头、智力低下,寿命可至成年 |
| Angelman 综合征 | 1/12000-1/20000 | 面孔似“快乐木偶”、 智力低下、肌张力低过度笑容、癫痫,寿命减少 |
| Prader–Willi 综合征 | 1/10000-1/30000 | 智力低下、肌张力低、性腺发育低下、肥胖、手足小、身材矮小,寿命减少 |
关注不罕见的“罕见”病【2】——Prader-Willi综合征
关注不罕见的“罕见”病【3】——Angelman综合征
贝比安Plus关注不罕见的“罕见”病【4】——猫叫综合征
贝比安Plus关注不罕见的“罕见”病【5】——1p36 deletion综合征
贝比安Plus关注不罕见的“罕见”病【6】——2q33.1 deletion综合征
贝比安Plus关注不罕见的“罕见”病【7】——Langer-Giedion 综合征
可以参考今年10.31的专题里的疾病介绍部分:http://www.berrygenomics.com/dna/
三、染色体单基因疾病
单基因遗传病是指由一对等位基因控制的疾病或病理性状。根据基因所处的染色体以及遗传方式的不同,可以分为常染色体显性遗传病、常染色体隐性遗传病、X伴性显性遗传病、X伴性隐性遗传病、Y伴性遗传病等几类。目前已经发现7600余种单基因病,中国人群中常见的单基因病包括遗传性耳聋、地中海贫血、进行性肌萎缩、成骨不全症和多囊肾等。
遗传性耳聋
聋病是影响人类健康和造成人类残疾的常见原因,许多聋病发病过程都与遗传相关。遗传性耳聋包括非综合征型耳聋和综合征型耳聋。研究表明,我国每年新增约6万先天性耳聋患者,至少50%的先天性耳聋由遗传因素导致,非综合征型耳聋占70%,约77%的非综合征型耳聋为常染色体隐性遗传。听力正常人群中约有6%为耳聋基因突变携带者,80%的耳聋患者都是由听力正常的夫妇所生育。约有0.3%的人群属于药物性耳聋敏感人群,7岁以下儿童因不合理使用抗生素造成耳聋的数量多达30万。而通过耳聋基因检测可为我国40%的聋病患者明确遗传学诊断。
为什么大多数遗传性耳聋由听力正常的父母生育呢?以常染色体隐性遗传耳聋为例。一对听力正常的父母,如果携带相同的耳聋基因,1/4的风险生育耳聋患者的后代,1/2的机会将耳聋基因传给孩子,但孩子听力正常,只有1/4的机会生育完全正常的孩子。
遗传性耳聋相关的热点基因及高频位点列表:| 基因 | 位点个数 | 临床症状 |
|---|---|---|
| GJB2 | 5 | 先天性重度以上感音神经性耳聋,50%先天性极重度听力损失患者与GJB2基因突变相关 |
| PDS | 11 | 大前庭水管综合征(EVAS),当患者患感冒、头部受打击时,听力急剧下降 |
| GJB3 | 2 | 后天高频感音神经性耳聋;表现为后天突发性耳聋 |
| 12S rRNA | 2 | 氨基糖甙类药物敏感性耳聋,患者使用氨基糖甙类药物会导致重度耳聋的发生,甚至发生“一针致聋” |
| KCNQ4 | 1 | 高频语后耳聋,该基因突变可导致渐进性高频听力下降,逐渐发展为全频率中-重度聋 |
| COCH | 1 | 多为高频神经性聋;该基因突变患者可能会出现一系列耳蜗、前庭功能障碍症状 |
| POU3F4 | 2 | 多为混合性耳聋;听力减退与年龄和频率有关 |
| GJB6 | 1 | 多为双侧中频至高频听力缺损,感音神经性耳聋,属于常染色体显性遗传非综合征耳聋 |
| TMIE | 1 | 属于常染色体隐性遗传非综合征耳聋,多表现为先天性重度以上感音神经性耳聋 |
脊髓性肌萎缩症(SMA)
脊髓性肌萎缩症(Spinal Muscular Atrophy, SMA)是一种常染色体隐性遗传的神经肌肉病,是由于脊髓前角细胞远动神经元变性,导致患者近端肌肉对称性、进行性萎缩和无力,最终导致呼吸衰竭甚至死亡。一旦SMA患儿出生,绝大部分在婴幼儿期就会发病,在人群中的携带率约为1/35-1/50左右,发病率约为1/6000-1/10000左右,目前尚无治疗手段,典型表现是进行性发展的肌肉萎缩,通常首先累及四肢,然后逐渐发展到躯干,造成患者各种运动功能严重受限,重症者终身与坐、站无缘,后期可能累及呼吸肌和吞咽肌,严重威胁患者的生命,呼吸衰竭和呼吸道感染是患者的主要死亡原因之一,对家庭来说不仅是经济上的重压,更是精神上的重压。
SMA通常根据疾病严重程度与发病年龄分为三个亚型。 I型患者(MIM#253300)在出生时或在6个月之前开始发病,不能独自坐或走,并且通常在两年内死于呼吸功能不全。II型患者(MIM# 253550)在6个月之后发病,可以独坐但在没有任何辅助设备帮助的情况下不能走路,并且寿命会大大减少,III型患者(MIM# 253400)通常在1岁半后发病,可以独立的坐或走路,但在青春期或成人后一般行走能力有所退步,需要靠轮椅行动。
我国目前有SMA携带者约3200万人,以每出生新生儿1600万计算,其中为携带者的准爸妈约80万,每年新增SMA患儿2500~3000人。据估计国内现存SMA患者约3~5万人,这是因为多数I、II型患者已于早期死亡。为了避免生出SMA患儿,准爸妈进行SMA携带筛查至关重要,在怀孕前或孕早期通过对准爸妈们简单的血液检测即可实现。
脊髓性肌萎缩症(SMA)是怎么遗传的?SMA是一种常染色体隐性遗传病,是由于5号染色体上的运动神经元存活基因1 (SurvivalMotor Neuron1,SMN1)纯合突变所致,其中95%为SMN1基因第7、第8外显子纯合缺失。
这个基因负责制造一种对运动神经元至关重要的蛋白,此基因的缺失会导致人体内缺少这种蛋白,引起的控制肌肉活动脊髓前角细胞(或称运动神经元)退化,造成肌肉无力及萎缩,这将严重影响患者的行动、呼吸和吞咽等功能,最终可能导致死亡。由于患儿父母通常各自只携带有一个SMN1缺陷基因,所以他们并不会患SMA,也不会出现疾病的症状和表征,这样的人群被称为携带者。而当子代同时从父母双方遗传到携带有缺陷的SMN1基因时,他们就将患SMA。
据目前全球流行病学的不完全统计,世界范围内大约每30-50人中就有一名SMA致病基因的携带者。常染色体隐性遗传病,两名携带者夫妇的后代可能是SMA患者、致病基因携带者与正常人中的任何一种。下图所示为SMA携带者夫妇生育后代时可能出现的基因组合。
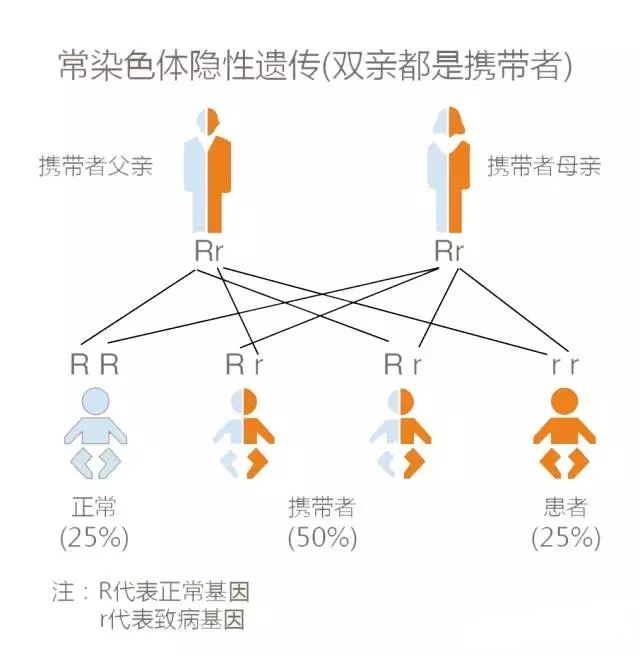
他们每次怀孕都有:25%的可能孩子会罹患SMA;50%的可能孩子会是SMA致病基因携带者;25%的可能孩子即不是SMA患者也不是携带者 。