
文献原文:
测序技术检测拷贝数变异在染色体疾病综合检测中的应用
Desheng Liang,* YingPeng,* Weigang Lv,* Linbei Deng,* Yanghui Zhang,* Haoxian Li,* Pu Yang,* Jianguang Zhang,† ZhuoSong,† Genming Xu,† DavidS. Cram,† and Lingqian Wu*
*医学遗传学国家重点实验室,中南大学,湖南长沙;†北京贝瑞和康生物技术有限公司,北京,中国
摘要:检测染色体拷贝数变异(copy number variation,CNV)在临床症状诊断不明和识别胎儿染色体疾病方面具有重要作用。目前,结合微阵列技术的染色体核型分析是临床检测 CNV的金标准。为了提高 CNV 检测的可行性并降低检测费用,我们推测第二代测序技术灵敏度和特异性与微阵列技术相当,可用于 CNV 检测。本研究同时采用中等密度单核苷酸多态性微阵列技术(single nucleotide polymorphism array,SNP array)和低覆盖度大规模平行测序技术(CNV sequencing, CNV-seq)对患者样本进行分析,配合 mate-pair 测序对 CNV-seq 检测到的 CNV 断裂点进行确认。CNV-seq 的最佳 DNA 检测样本量为 50 ng,而样本量低至 10 ng 也可实现准确的 CNV 检测。对微小 CNV 样本进行验证研究,结果显示 CNV-seq 技术具有良好的特异性及可重复性,其检测分辨率约为 0.1Mb。对 72 个已由 SNP Array 检测过的样本进行 CNV-seq 盲测,发现 CNV-seq 与 SNP Array 具有较高的检测一致性。由此认为,CNV-seq 可作为微阵列技术的替代方法用于染色体疾病检测。(J Mol Diagn2014,16:519e526;http://dx.doi.org/10.1016/j.jmoldx.2014.05.002)
人类已知的染色体疾病有200多种1。大多数染色体疾病由染色体数目异常引起,以唐氏综合征最为普遍。另有部分染色体疾病因缺失或重复一段染色体片段(拷贝数变异,copy number variations,CNVs)而引起,统称为染色体微缺失/微重复综合征。
染色体疾病临床表型多样,表现为多发性先天畸形、肢体残疾、发育迟缓、智力障碍、癫痫症、自闭症和学习障碍等1-3。近期对人类配子和植入前胚胎的研究揭示4-6,患者固有的染色体不稳定性是其染色体发生异常的主要原因。CNV的形成与众所周知的非同源末端连接,以及新近提出的DNA 复制扰动和不连续 DNA 复制有关。多种机制共同作用,原发性CNV的形成速度远远超过其他类型的基因变异7。同时一些发生率低、但影响显著的染色体变异持续通过家族遗传得以传播1。通常,大多数原发性或继承性的染色体异常发生时,胎儿仍可发育至足月,导致约0.3%的染色体疾病患儿出生8。
过去 30 年,产前诊断对早、中孕期的染色体异常检测起关键作用,选择性终止妊娠可减少新生儿中染色体疾病的发病率1。产前诊断需针对胎儿进行一系列分析,包括母体血清学筛查和超声检测。对于疑似染色体疾病的胎儿,后续诊断通过绒毛膜取样或羊膜穿刺术进行核型分析。核型分析检测超过20个的细胞分裂中期细胞,分辨率约为5Mb,是目前检测胎儿染色体非整倍体、多倍体、平衡和非平衡性结构重排、较大片段的微缺失/微重复,以及嵌合体的金标准9-11。其他方法如荧光原位杂交(fluorescence in situ hybridization,FISH)12、荧光定量聚合酶链式反应(polymerase chain reaction,PCR)13和多重连接探针扩增技术14(multiplex ligation-dependent probe amplification,MLPA)也被用于胎儿染色体非整倍体的快速检测。最近,高分辨率寡核苷酸和单核苷酸多态性微阵列技术(single nucleotide polymorphism array,SNP array)也被用于产前诊断,并为其带来巨大变革15。与其他技术相比,SNP array 能在更广范围内进行染色体异常的检测,可发现具有临床意义的、小于 5 Mb 的染色体微缺失/微重复16。
过去 30 年,产前诊断对早、中孕期的染色体异常检测起关键作用,选择性终止妊娠可减少新生儿中染色体疾病的发病率1。产前诊断需针对胎儿进行一系列分析,包括母体血清学筛查和超声检测。对于疑似染色体疾病的胎儿,后续诊断通过绒毛膜取样或羊膜穿刺术进行核型分析。核型分析检测超过20个的细胞分裂中期细胞,分辨率约为5Mb,是目前检测胎儿染色体非整倍体、多倍体、平衡和非平衡性结构重排、较大片段的微缺失/微重复,以及嵌合体的金标准9-11。其他方法如荧光原位杂交(fluorescence in situ hybridization,FISH)12、荧光定量聚合酶链式反应(polymerase chain reaction,PCR)13和多重连接探针扩增技术14(multiplex ligation-dependent probe amplification,MLPA)也被用于胎儿染色体非整倍体的快速检测。最近,高分辨率寡核苷酸和单核苷酸多态性微阵列技术(single nucleotide polymorphism array,SNP array)也被用于产前诊断,并为其带来巨大变革15。与其他技术相比,SNP array 能在更广范围内进行染色体异常的检测,可发现具有临床意义的、小于 5 Mb 的染色体微缺失/微重复16。
在临床应用中,微阵列芯片通常需定制,采用高密度寡核苷酸或 SNP 探针均匀覆盖每条染色体以及致病基因的外显子区域3,16,17。结合双寡核苷酸的 SNP array 也被用于染色体分析,其分辨率更高,可额外检测出寡核苷酸微阵列未能检测到的、具有临床意义的染色体异常18,19。此外,根据公共数据库中详尽的染色体异常及相关临床表型信息,使用定制微阵列芯片检测 CNVs 易于实现准确的临床检测20,可对更加复杂的综合征(如自闭症)进行深入的遗传基础研究21。然而,对某些罕见的、数据库中未涵盖的CNVs,仍无法实现可靠的诊断22。相比核型分析,基因微阵列技术能够额外检测到 5% ~ 15% 的染色体异常16,23,但在检测多倍体和平衡易位方面,基因微阵列技术仍存在难度。一项关于低危和高危妊娠的研究显示,微阵列可作为检测胎儿染色体异常的主要技术加以运用24。
在西方国家,联合应用超声、核型分析以及微阵列技术已对识别妊娠期胎儿染色体异常、诠释习惯性流产,以及诊断儿童和成人未知生理和心理问题产生重大影响,可为提高其生活质量提供更好的治疗方案16,25,26。但在发展中国家和一些发达国家,微阵列技术因技术难度高、缺少专业知识和高成本等尚未得到广泛运用,故而新生儿染色体疾病发病率仍很高27-29。目前,临床上亟需微阵列技术的替代方法,以便全面准确、经济实惠地检测大多数染色体疾病。我们推测基于第二代测序技术的CNV-seq有望满足这一需求。
我们之前的研究结果显示采用低覆盖度鸟枪测序法分析约500万条的测序序列,以每条染色体上连续的20kb基因组为测序单元(bin),可检测5%的X染色体嵌合体30。我们推测这一方法同样适用于22对常染色体以及 X、Y两条性染色体的高分辨率 CNV 检测。本研究采用CNV-seq检测受检样本,其染色体异常均由SNP array确认,结果表明,CNV-seq 与 SNP array 具有高度的检测一致性,且CNV-seq重复性好、灵敏度高,分辨率约 0.1 Mb。
材料与方法
受检样本
核型分析和SNP array在中南大学医学遗传学国家重点实验室和湖南家辉遗传专科医院进行。入选样本共72例,62例来自于发育迟缓、智力障碍或先天畸形患者,10例来自于流产组织,详细情况见补充材料(表S1)。另有3例环状染色体样本和4例微小CNVs样本(小于 0.25 Mb)用于评估 CNV-seq的检测灵敏度和特异性。使用Qiagen公司生产的 DNeasy Blood & Tissue Kit试剂盒,提取患者血液和流产组织中的DNA,琼脂糖凝胶电泳后,使用Thermo Fisher Scientific公司生产的 NanoDrop 分光光度计,评估 DNA 样品的质量和浓度。
SNP array
使用 Illumina 公司生产的 HumanCytoSNP-12 BeadChip芯片进行染色体 CNV 分析,SNP 探针密度为 298,563,基因组平均间距为 19 kb。使用 Illumina 公司的 GenomeStudio 软件(版本 2011.1)计算 log R 值以及 A和 B等位基因频率值。使用Illumina 公司的 cnvPartiion 软件插件(版本 v3.1.6)进行详细的 CNV 分析。CNV重复(AAA,AAB,ABB 和 BBB等位基因组合)和缺失(A或 B等位基因)定义为50 个 SNP 区域中置信度得分 >100。
CNV-seq
将 50 ng 基因组 DNA 进行片段化处理,获得平均大小为 300 bp 的DNA片段,参照文献方法构建测序文库31,32,使用 Illumina 公司的 HiSeq 2000平台进行测序,36bp单端测序,测序深度0.1倍,产生800万条的测序序列。使用 Burrows-Wheeler 算法将所有测序序列与hg19基因组进行比对分析33。根据文献记载的数据处理和分析算法30,将最少 20 个测试样本进行内部比较,互相作为参考。为了提高检测灵敏度,以60Kb为基本测序单元,对大约500万条测序序列进行分析。以标准化测序读取密度的 log2 值为y轴,以相对连续的60kb测序单元为x轴,绘制CNV-seq检测结果图。然后,依据每条染色体的长度计算 log2 平均值。染色体拷贝数重复(3个拷贝)的log2 理论值为log2 [1.5] = 0.58;染色体拷贝数缺失(1个拷贝)的log2 理论值为log2 [0.5] = -1.0。将CNV-seq检测拷贝数重复的cut-off值设为 >2.8(log2 [1.4] = 0.49),拷贝数缺失的cut-off值设为 <1.2(log2 [0.6] = 0.74)。
Mate-pair 测序
使用 Illumina 公司的 Nextera Mate-Pair Sample Preparation Kit 进行 mate-pair 测序。先将5µg基因组 DNA 进行片段化处理,然后将DNA片段溶于1%的琼脂糖凝胶,再用 Qiagen 公司的 QIAquick Gel Extraction Kit 对 5 kb 大小的 DNA 组分进行纯化。使用 HiSeq 2000对文库进行测序,产生成对的正反向约为 100 bp 的测序序列,然后使用 Burrows-Wheeler 算法将这些测序序列唯一比对到 hg19 参照基因组中33。共有10-2,500 万个测序序列用来识别至少一个成对的染色体断裂点。
结果与分析
CNV-seq 对低样本量的检测效果
本研究小组在以往的研究中证实,当用于构建文库的初始DNA量为100ng时,CNV-seq可取得与核型分析一致的检测结果30。考虑到临床样本DNA量的可变性,本研究对正常样本(46,XX)和Wolf-Hirschhorn syndrome样本(46,XX,4p16.1-pter;8.92Mb缺失)进行低样本量的CNV-seq检测,以验证CNV-seq的临床适用性。分别重复46,XX和46,XX,4p16.1-pter样本量为10ng和50ng的CNV-seq检测(图1,46,XX和46,XX,4p16.1-pter样本量分别为10ng和50ng的4号染色体CNV-seq结果图;补充材料图S1和S2,46,XX,4p16.1-pter样本量为50ng时所有染色体的 CNV-seq结果图)。样本量为50ng时,CNV-seq对46,XX样本和46,XX,4p16.1-pter样本的正常染色体检测均未发现显著的CNV,46,XX,4p16.1-pter样本也在4p16.1-pter区段观察到预期的拷贝数缺失。与之相反,样本量为10ng时,46,XX正常样本或46,XX,4p16.1-pter异常样本的CNV-seq结果图表现出轻微的不稳定性,部分染色体表现出轻微的非特异性 CNV 波动,染色体末端区域表现显著。而在46,XX,4p16.1-pter异常样本中,还观察到比 4p16.1-pter 区域稍小的拷贝数缺失。基于以上分析,50ng为最佳的 CNV-seq检测样本量,可产生 800 万条测序序列。
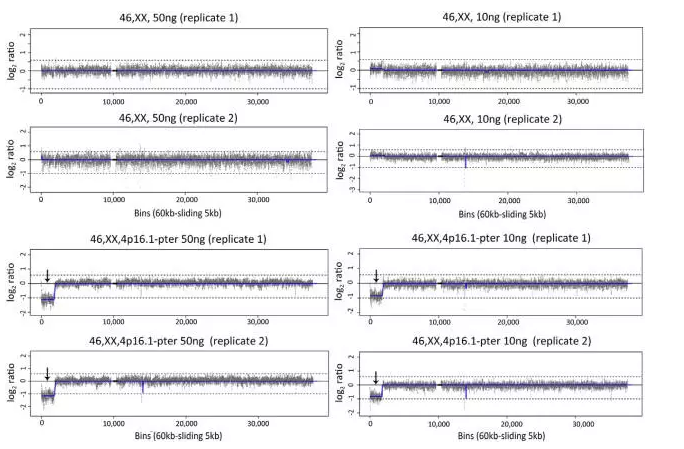
图 1 CNV-seq 对低样本量的检测效果。图为 46,XX和46,XX,4p16.1-pter样本量分别为10ng和50ng的4号染色体CNV-seq结果图。CNV-seq 结果图以标准化测序读取密度的 log2 值为y 轴,以相对连续的60 kb 测序单元为x 轴。上虚线 [log2 (3/2)] 和下虚线[log2 (1/2)] 分别表示100%染色体增加(重复)和100%染色体减少(缺失)。CNV区域、重复序列区域和着丝粒区域分别用蓝色线条、红色方框和黑色方框表示。箭头对应4p16.1-pter区域8.92 Mb的缺失,50 ng样本量检测效果更佳。
CNV-seq 的特异性和可重复性
在对样本的检测中,CNV-seq偶尔检出一些大小为几百Kb的微缺失/微重复。为了验证这些微小 CNVs 是否真实存在,以及CNV-seq 对真正CNVs检测的特异性,我们使用 mate-pair 测序来确认和精确定位两个已测样本的缺失和断裂位点。其中一个样本Xp22.2区段缺失0.22 Mb,可能导致 PLP1 基因缺失和 X 连锁佩梅病(Pelizaeus-Merzbacher disease);另一个样本6q26区段纯合性缺失0.08Mb,可能与帕金森病(Parkinson disease)PARK2基因缺失有关。mate-pair 测序至少检测出两个样本跨越断裂点的5kb DNA片段(图2和图3)。基于上述 mate-pair 测序结果,设计引物扩增包含两个断裂点的小片段 DNA,运用Sanger测序法将疑似基因断裂点精确定位到单核苷酸水平,结果证实,最初由 CNV-seq 检测到的两个缺失真正存在。
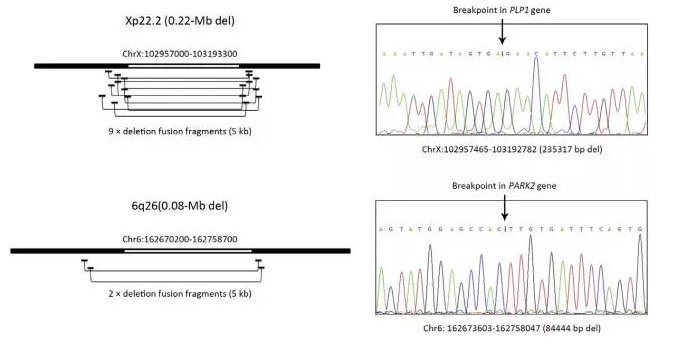
图2 Mate-pair 测序确认由 CNV-seq 识别出的两个缺失的断裂点(Xp22.2区段缺失0.22 Mb和6q26区段缺失0.08 Mb)。根据横跨缺失区域的 5 kb 片段 mate-pair 测序结果,以 hg19 作为参考基因组定义缺失区域的近似坐标,并在断裂点的任一侧设计 PCR 引物。Sanger 测序法确定缺失片段大小,并将断裂点定位到单核苷酸(箭头)水平。由此确认,X 连锁佩梅病和帕金森病疑似患者中的 Xp22.2 区段和 6q26区段缺失分别由 PLP1和 PARK2基因序列的全部或部分缺失引起。
为了确定能否重复检测上述两种微小 CNVs,我们使用3份50 ng 基因组DNA重复检测。结果发现,三次mate-pair 测序均可发现Xp22.2区段0.22Mb的缺失和6q26区段0.08Mb的缺失,且CNV缺失的拷贝数也符合预期(图3)。后续分析另外两个基因组 DNA 样本,第一个样本显示22q11.2 区段缺失0.24 Mb,查询OMIM数据库显示,缺失导致3个非致病基因(IGLL3P,LRP5L 和 CRYBB2P1)半合子表达。第二个样本显示9q33.1区段缺失0.22 Mb,查询OMIM数据库显示,缺失导致致病基因TRIM32半合子表达。在两个DNA样本的预期位置均重复检测到单个CNV(数据未示出)。由此可知应用CNV-seq检测微小 CNVs 具有良好的特异性和重复性。
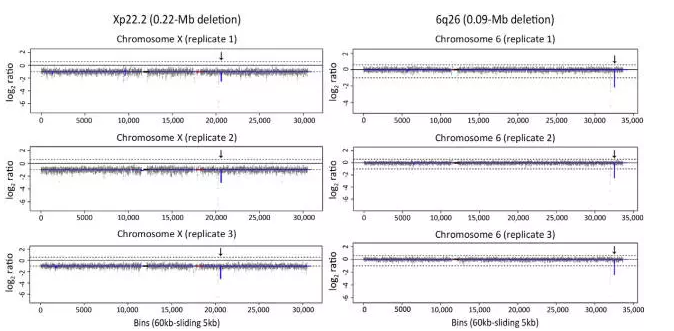
图3 CNV-seq 对微小 CNV 的重复性试验。三个重复样本的 CNV 结果图均显示Xp22.2区段0.22Mb的缺失和6q26区段0.08Mb的缺失。CNV-seq 结果图以标准化测序读取密度的 log2 值为y 轴,以相对连续的60 kb 测序单元为x 轴。上虚线 [log2 (3/2)] 和下虚线[log2 (1/2)] 分别表示100%染色体增加(复制)和100%染色体减少(缺失)。CNV区域、重复序列区域和着丝粒区域分别用蓝色线条、红色方框和黑色方框表示。三个重复样本中的两个缺失(箭头)均被 CNV-seq 重复检测到,且CNV倍性符合预期。
验证CNV-seq 检测染色体疾病的临床适用性
为了评估 CNV-seq 在染色体疾病综合诊断中的适用性,我们对72例经由SNP array确认过的样本进行CNV-seq分析(个别样本的保存期已超过 5 年)。检测过程为双盲检测,CNV-seq 结果与SNP array 结果进行比较(数据概况见表1,完整数据见补充表S1)。文章分析了11例样本的CNV-seq和SNP array比较结果,其中4例见图4,另外7例见补充图 S3。CNV-seq对 72 例样本的检测结果与SNP array完全一致(表 1 和补充表 S1),其中致病CNVs43例(60%)。此外,CNV-seq 还检测到5例未被SNP array 检出的小于 1 Mb 的次级 CNVs(0.20 ~ 0.6 Mb),其临床意义未知。在所有诊断一致的样本中,染色体微缺失/微重复的性质、大小以及染色体异常的位置, SNP array 和 CNV-seq 结果几乎相同,也与核型分析结果基本一致。上述结果表明,CNV-seq 可做为染色体疾病的检测方法,其检测效果与 SNP array相当。
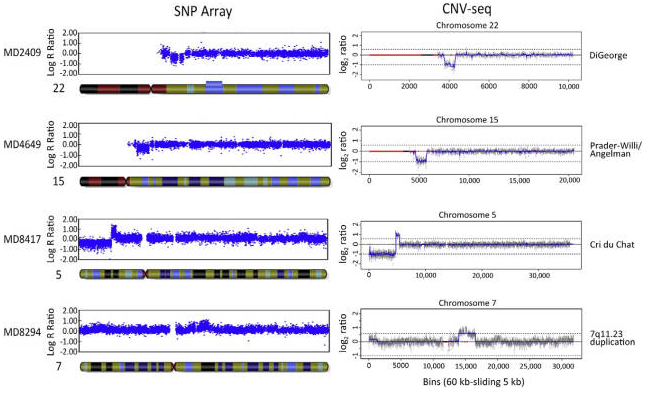
图 4 比较 SNP array 和 CNV-seq对四种染色体疾病的检测结果。两种方法测定的染色体位置和 CNVs 大小基本相同。各个 CNV 的详细信息见补充表 S1。
表 1 比较SNP array 和 CNV-seq 对染色体疾病的检测一致性
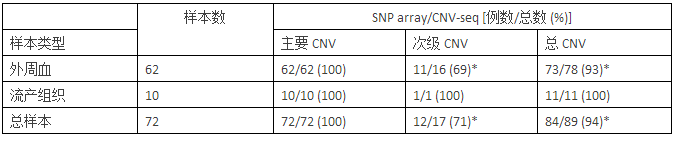
*注:由于HumanCytoSNP-12 BeadChip微阵列芯片检测的不稳定性,5例次级CNV未被SNP array检出。
CNV-seq 检测环状染色体缺失
为了验证 CNV-seq 是否能够检测其他类型的染色体异常,CNV-seq重新分析一组经由核型分析证实具有环状染色体结构(14号、22号和 18 号染色体,图 5)的样本。CNV-seq 分析发现14号染色体q 端缺失3.2 Mb,22号染色体q端缺失7Mb,18号染色体p端缺失2.5 Mb ,q端缺失15.7 Mb(图5),上述缺失是环状染色体形成过程中随机末端缺失导致的结果。对于 18 号环状染色体,CNV-seq 清楚地检测到两个符合预期的p端和q端缺失,缺失的大小也被精确识别,映射环状染色体的形成过程。不过CNV-seq 不能映射 14p 和 22p的缺失。
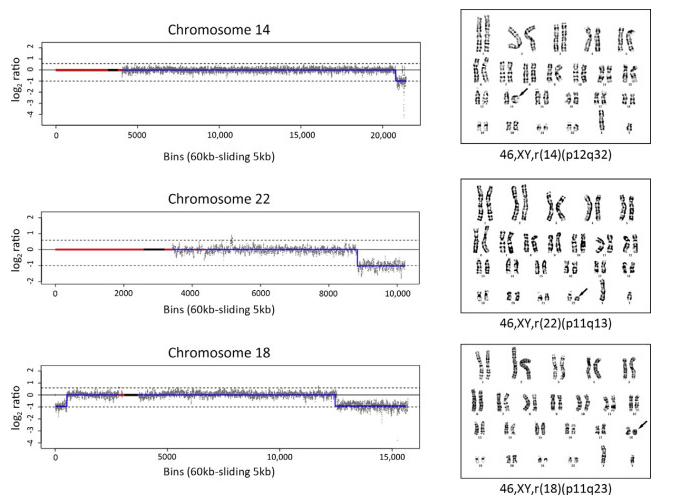
图 5 CNV-seq 对14号、22号和 18 号环状染色体的检测结果。图中显示 CNV-seq 和匹配的核型分析结果。CNV-seq 结果图以标准化测序读取密度的 log2 值为y 轴,以相对连续的60 kb 测序单元为x 轴。上虚线 [log2 (3/2)] 和下虚线[log2 (1/2)] 分别表示100%染色体增加(复制)和100%染色体减少(缺失)。CNV区域、重复序列区域和着丝粒区域分别用蓝色线条、红色方框和黑色方框表示。CNV-seq 可精确映射和测量终端断裂点。
讨论
本研究评估了CNV-seq对染色体疾病检测的可靠性和准确性。通过检测 72 例经由 SNP array 确诊的样本,发现 CNV-seq 可取得与 SNP array 完全一致的检测结果。同时,CNV-seq 检测到5例未被 SNP array检出的次级 CNVs(<1Mb)。对这5例次级CNVs分析发现,CNV 区间缺少可供分析的 SNPs 导致SNP array未能检出。总体而言,CNV-seq 可准确检测全部或部分染色体非整倍体、微缺失/微重复(<1 Mb),以及环状染色体。此外,CNV-seq 可检测 0.1 Mb 的杂合性和纯合性缺失,且这些微小缺失经mate-pair 测序确认真实存在。由此可知,CNV-seq 可用于检测具有临床意义的染色体疾病,且具有高度的灵敏度和特异性。
CNV-seq 需在基因组覆盖度和分辨率之间实现平衡,且检测染色体疾病需经济有效。为此,我们将测序序列产出设定为 500 万条,同时以连续的60 kb 为基本测序单元,每条序列的平均读长为 150 ~ 165bp。尽管相同或不同样本间每个测序单元读取的序列具有随机性,但CNV-seq 仍能精确检测一系列具有临床意义的 CNVs。虽然有必要对 CNV-seq 进行全基因组水平验证,但本研究表明,CNV-seq可能适用于整个基因组范围的 CNV 分析(p 臂近着丝粒的高度重复区域、Y染色体q端异染色质区域、着丝粒序列以及其他包含重复序列的区域除外)16。此外,CNV-seq 检测获得的 CNV 区域坐标和 SNP array结果高度一致。
由于 CNV-seq 的高性能,我们推测第二代测序技术更优于SNP array。本研究还发现,CNV-seq 可检测出与 X 连锁佩梅病相关的 PLP1基因 0.22 Mb 的缺失,以及与帕金森病相关的 PARK2 基因0.08 Mb 的纯合性缺失。因此,CNV-seq 有可能发现0.1 Mb 缺失引起的常染色体隐性遗传、X 连锁和常染色体显性遗传疾病。另一方面,寡核苷酸微阵列芯片可根据已知致病基因的外显子区域设计探针,对涉及一个或多个外显子缺失的遗传病进行单基因检测,检测范围可从染色体疾病扩展至单基因病16,19。另外,SNP array 可根据特异性点突变、微小缺失、插入等设计SNP 探针,进一步扩大其检测单基因病的效能17,26。不过目前尚无单一的微阵列平台用于临床染色体疾病和单基因病的综合检测。对于已知家族病史的单基因病检测,除可沿用标准的 PCR 方法外,也可使用二代测序技术进行全外显子组检测35。相较于寡核苷酸微阵列和 CNV-seq,SNP array 还可通过一系列连续的 SNP 探针检测单亲二倍体和判断血缘关系16。单亲二倍体在新生儿中的发病率为 0.03%36,最近有研究结合寡核苷酸和 SNP 平台进行单亲二倍体检测18,19。
CNV-seq 可依据数据分析对 CNV实现量化。本研究CNV-seq识别的所有染色体重复和缺失拷贝数均值 ± 标准误差分别为 3.0 ± 0.1 和 1.0 ± 0.1,有潜力测定其他具有临床意义的CNVs。以往研究发现CNV-seq 可检测性染色体母体嵌合30和胎盘嵌合37,38,本研究进一步证实上述发现。随着对嵌合体认识的日益加深39,CNV-seq 可能成检测嵌合体的有效工具,更好诠释基因型与表型的关联。此外,滋养外胚层囊胚活检和全基因组扩增技术越来越多地被用于胚胎植入前遗传学诊断40,41,CNV-seq 也可特异的检测与滋养外胚层细胞相关的低水平嵌合,42。另外,CNV-seq可精确检测低至10ng的样本,比微阵列技术更具临床适用性,可用于分析低样本量的临床样本和宝贵的科研样本,而微阵列技术所需的最低样本量则是其 20 倍。
在许多发展中国家,如印度和中国,产前诊断仍主要依赖于胎儿染色体核型分析、母血清学筛检和超声检测进行。无创性产前检测32的引进以及高危和低危孕妇群体的增加正逐渐改变这一现状,现已对早期染色体非整倍体的检测产生影响。然而,产后新生儿检查仍主要依赖表型,很少进行身体以及智力方面的评估。微阵列技术由于普及性差、价格高昂,仅限于小部分人作为辅助手段进行产前和产后检测。因此,新生儿和成人染色体疾病带来的社会和经济负担居高不下27-29。本研究检验了CNV-seq在染色体疾病检测方面的效能。结果表明,广泛推行的新一代测序技术有望显著降低染色体疾病在发达国家和发展中国家的发病率。首先,CNV-seq 能检出大约 0.1 Mb CNVs 引发的已知染色体疾病,故可用于分析各类型样本,包括羊水、绒毛、流产组织和外周血等;其次,测序试剂的成本将随着时间的推移而显著降低,加之工作流程简单,CNV-seq 有望成为比微阵列技术更具扩展性且经济实惠的染色体疾病检测替代技术。此外,借助于更深层次的测序,CNV-seq 有望得以改进,用于高分辨率的 CNV 和 SNP 分析,更全面地检测遗传性疾病。
参考文献:
1. Gardner RJM,Sutherland GR, Shaffer LG (Eds): Chromosome Ab- normalities and Genetic Counselling. New York, Oxford University Press, 2011
2. Breman A,Pursley AN, Hixson P, Bi W, Ward P, Bacino CA, Shaw C, Lupski JR, Beaudet A,Patel A, Cheung SW, Van den Veyver I: Prenatal chromosomal microarray analysisin a diagnostic laboratory: experience with >1000 cases and review of the literature. Prenat Diagn 2012, 32:351e361
3. Stankiewicz P,Beaudet AL: Use of array CGH in the evaluation of dysmorphology, malformations,developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev2007, 17:182e192
4. Nagaoka SI,Hassold TJ, Hunt PA: Human aneuploidy: mechanisms and new insights into anage-old problem. Nat Rev Genet 2012, 13: 493e504
5. VoetT,VannesteE,VermeeschJR:The human cleavage stage embryo is a cradle of chromosomal rearrangements. Cytogenet Genome Res 2011,133:160e168
6. Vanneste E, VoetT, Le Caignec C, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M,Schuit F, Fryns JP, Verbeke G, D’Hooghe T, Moreau Y, Vermeesch JR: Chromosome instability is common in human cleavage-stage embryos. Nat Med 2009, 15:577e583
7. Hastings PJ,Lupski JR, Rosenberg SM, Ira G: Mechanisms of changes in gene copy number. NatRev Genet 2009, 10:551e564
8. Hassold T, HuntP: To err (meiotically) is human: the genesis of human aneuploidy. Nat RevGenet 2001, 2:280e291
9. Bayani J, SquireJA: Traditional banding of chromosomes for cyto- genetic analysis. Curr ProtocCell Biol 2004. Chapter 22:Unit 22
10. Smeets DF: Historical prospective of human cytogenetics: from mi- croscope to microarray. Clin Biochem 2004,37:439e446
11. Trask BJ: Human cytogenetics: 46 chromosomes, 46years and counting. Nat Rev Genet 2002, 3:769e778
12. Jia CW, Wang SY, Ma YM, Lan YL, Si YM, Yu L, ZhouLY: Fluorescence in situ hybridization in uncultured amniocytes for detection of aneuploidy in 4210 prenatal cases. Chin Med J(Engl) 2011, 124:1164e1168
13. Mann K, Hills A, Donaghue C, Thomas H, Ogilvie CM:Quantitative fluorescence PCR analysis of >40,000 prenatal samples for therapid diagnosis of trisomies 13, 18 and 21 and monosomy X. Prenat Diagn 2012,32:1197e1204
14. Van Opstal D, Boter M, de Jong D, van den Berg C,Bruggenwirth HT, Wildschut HI, de Klein A, Galjaard RJ: Rapid aneuploidy detection with multiplex ligation-dependent probe amplification: a prospectivestudy of 4000 amniotic fluid samples. Eur J Hum Genet 2009, 17:112e121
15. Shaffer LG, Rosenfeld JA: Microarray-based prenatal diagnosis for the identification of fetal chromosome abnormalities. Expert RevMol Diagn 2013, 13:601e611
16. Schaaf CP, Wiszniewska J, Beaudet AL: Copy numberand SNP arrays in clinical diagnostics. Annu Rev Genomics Hum Genet 2011, 12:25e51
17. Keren B, Le Caignec C: Oligonucleotide microarraysin constitutional genetic diagnosis. Expert Rev Mol Diagn 2011, 11:521e532
18. Fan YS, Ouyang X, Peng J, Sacharow S, Tekin M,Barbouth D, Bodamer O, Yusupov R, Navarrete C, Heller AH, Pena SDj: Frequentdetection of parental consanguinity in children with developmental disorders bya combined CGH and SNP microarray. Mol Cytogenet 2013, 6:38
19. Wiszniewska J, Bi W, Shaw C, Stankiewicz P, KangSH, Pursley AN, Lalani S, Hixson P, Gambin T, Tsai CH, Bock HG, Descartes M,Probst FJ, Scaglia F, Beaudet AL, Lupski JR, Eng C, Wai Cheung S, Bacino C,Patel A: Combined array CGH plus SNP genome analyses in a single assay foroptimized clinical testing. Eur J Hum Genet 2014, 22:79e87
20. Howard HJ, Beaudet A, Gil-da-Silva Lopes V, Lyne M,Suthers G, Van den Akker P, Wertheim-Tysarowska K, Willems P, Macrae F:Disease-specific databases: why we need them and some recommen- dations fromthe Human Variome Project Meeting, May 28, 2011. Am J Med Genet A 2012,158A:2763e2766
21. Celestino-Soper PB, Shaw CA, Sanders SJ, Li J,Murtha MT, Ercan- Sencicek AG, Davis L, Thomson S, Gambin T, Chinault AC, Ou Z,German JR, Milosavljevic A, Sutcliffe JS, Cook EH Jr, Stankiewicz P, State MW,Beaudet AL: Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet2011, 20:4360e4370
22. Klopocki E, Mundlos S: Copy-number variations,noncoding se- quences, and human phenotypes. Annu Rev Genomics Hum Genet 2011,12:53e72
23. Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM,Zachary JM, Savage M, Platt LD, Saltzman D, Grobman WA, Klugman S, Scholl T,Simpson JL, McCall K, Aggarwal VS, Bunke B, Nahum O, Patel A, Lamb AN, Thom EA,Beaudet AL, Ledbetter DH, Shaffer LG, Jackson L: Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012, 367:2175e2184
24. Fiorentino F, Napoletano S, Caiazzo F, Sessa M,Bono S, Spizzichino L, Gordon A, Nuccitelli A, Rizzo G, Baldi M:
25. Chromosomal microarray analysis as a first-linetest in pregnancies with a priori low risk for the detection of submicroscopic chromosomal abnormalities. Eur J Hum Genet 2013, 21:725e730
26. Callaway JL, Shaffer LG, Chitty LS, Rosenfeld JA,Crolla JA: The clinical utility of microarray technologies applied to prenatalcytoge- netics in the presence of a normal conventional karyotype: a review ofthe literature. Prenat Diagn 2013, 33:1119e1123
27. Schwartz S: Clinical utility of single nucleotide polymorphism arrays. Clin Lab Med 2011, 31:581e594
28. Bian XM, Guo Q, Qi QW: Current situation and development of prenatal diagnosis in China. Front Med China 2010, 4:271e27428. Kaur A, Singh JR: Chromosomal abnormalities: genetic disease burden
29. in India. Int J Hum Genet 2010, 10:1e1429. Verma IC: Burden of genetic disorders in India. Indian J Pediatr 2000,
30. 67:893e89830. WangY,ChenY,TianF,ZhangJ,SongZ,WuY,HanX,HuW,
31. Ma D, Cram D, Cheng W: Maternal mosaicism is asignificant contributor to discordant sex chromosomal aneuploidies associatedwith noninvasive prenatal testing. Clin Chem 2014, 60:251e259
32. LiangD,LvW,WangH,XuL,LiuJ,LiH,HuL,PengY,WuL:Non-invasive prenatal testing of fetal whole chromosome aneuploidy by massively parallel sequencing. Prenat Diagn 2013, 33:409e415
33. Song Y, Liu C, Qi H, Zhang Y, Bian X, Liu J:Noninvasive prenatal testing of fetal aneuploidies by massively parallel sequencing in a prospective Chinese population. Prenat Diagn 2013, 33:700e706
34. Li H, Durbin R: Fast and accurate short read alignmentwith Burrows- Wheeler transform. Bioinformatics 2009, 25:1754e1760
35. Asan, Geng C, Chen Y, Wu K, Cai Q, Wang Y, Lang Y,Cao H, Yang H, Wang J, Zhang X: Paired-end sequencing of long-range DNAfragments for de novo assembly of large, complex Mammalian ge- nomes by directintra-molecule ligation. PLoS One 2012, 7:e46211
36. Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A,Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, BekheirniaMR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR,Beaudet AL, Gibbs RA, Eng CM: Clinical whole-exome sequencing for the diagnosisof mendelian dis- orders. N Engl J Med 2013, 369:1502e1511
37. Liehr T: Cytogenetic contribution to uniparental disomy (UPD). Mol Cytogenet 2010, 3:8
38. WangY,ZhuJ,ChenY,LuS,ChenB,ZhaoX,WuY,HanX,MaD, LiuZ, Cram D, Cheng W: Two cases of placental T21 mosaicism: challenging thedetection limits of non-invasive prenatal testing. Prenat Diagn 2013,33:1207e1210
39. Mao J, Wang T, Wang BJ, Liu YH, Li H, Zhang J, CramD, Chen Y: Confined placental origin of the circulating cell free fetal DNArevealed by a discordant non-invasive prenatal test result in a trisomy 18pregnancy. Clin Chim Acta 2014, 433C:190e193
40. Biesecker LG, Spinner NB: A genomic view of mosaicism and human disease. Nat Rev Genet 2013, 14:307e320
41. Franasiak JM, Forman EJ, Hong KH, Werner MD, UphamKM, Treff NR, Scott RT Jr: The nature of aneuploidy with increasing age of thefemale partner: a review of 15,169 consecutive trophectoderm bi- opsiesevaluated with comprehensive chromosomal screening. Fertil Steril 2013,101:656e663.e1
42. Schoolcraft WB, Katz-Jaffe MG: Comprehensive chromosome screening of trophectoderm with vitrification facilitates electivesingle- embryo transfer for infertile women with advanced maternal age. FertilSteril 2013, 100:615e619
43. Liu J, Wang W, Sun X, Liu L, Jin H, Li M, Witz C,Williams D, Griffith J, Skorupski J, Haddad G, Gill J: DNA microarray revealsthat high proportions of human blastocysts from women of advanced maternal ageare aneuploid and mosaic. Biol Reprod 2012, 87:148. 1e9

